Liver Transplantation for Propionic Acidemia:
Part 1 – Answers to Questions that Families May Have
James Squires, MD, MS

Dr. James Squires
Dr. Squires is a liver disease specialist at Children’s Hospital of Pittsburgh of UPMC and an assistant professor of pediatrics at the University of Pittsburgh School of Medicine.
Jodie M. Vento, MGC, LCGC
Jodie Vento is a genetic counselor and manager of the Center for Rare Disease Therapy at the Children’s Hospital of Pittsburgh of UPMC.
What can we expect that a liver transplant could do for our child?
Based on experience to date with liver transplants in children with Propionic Acidemia (PA),we can say that after a liver transplant,children are likely to have a substantially better quality of life and a dramatic reduction in metabolic crises. It’s important for families to understand, however, that liver transplantation is not a cure for PA. This is because the enzyme deficiency that causes PA exists throughout the body, not just in the liver.
The liver transplant serves as what we liver specialists call a bulk enzyme replacement, providing enough functional enzyme to minimize – if not eliminate –metabolic crises, which are the most severe complications of PA for affected children as well as one of the most frightening features of the disease for families.
Because complications related to PA may still occur following a transplant, there will be a continued need for your child to get follow-up care with one or more medical specialists.
Is there a minimum or “best” age for a child with PA to have a liver transplant?
There is no minimum or “best” age. At our center, the average age of a liver transplant for a child with PA is about seven years old, but we have performed transplants in children as young as one year old.
The best time to consider a liver transplant is while the symptoms of PA are still reasonably well controlled. There is also no minimum age for undergoing a pre-transplant evaluation or being placed on the transplant waiting list.
What should we consider when deciding where to take our child for a liver transplant evaluation?
The most important factor to consider is the experience of the surgical team performing liver transplants in patients with PA and other metabolic diseases. These patients have complex needs that are different from those of patients receiving liver transplants for other conditions.
The pediatric liver transplantation program at Children’s Hospital of Pittsburgh of UPMC was established in 1981 by world-renowned transplant surgeon Thomas E. Starzl, MD, PhD. Our t Director of Pediatric Transplantation, George Mazariegos, MD, FACS, pioneered liver transplantation for children with metabolic diseases in 2004. Since that time, Children’s Hospital has performed more than 330 liver transplants for children with metabolic disease,more than any other transplant center.
We’ve also performed more liver transplants in children than any other center in the United States and more living-donor transplants than any other pediatric center in the country. Our one-year survival rate for pediatric liver transplant patients is 98%, exceeding the national average of 95%, according to the Scientific Registry of Transplant Recipients, Jan. 2018 release.
In addition to our world-renowned and experienced liver transplant surgeons, our Center for Rare Disease Therapy includes international experts in the diagnosis and treatment of PA and other metabolic diseases.
How would we start the process of having our child evaluated for a liver transplant?
I can tell you how the process works here at Children’s Hospital of Pittsburgh of UPMC. It starts with a referral from your doctor or hospital requesting that we evaluate your child. We also receive self-referrals directly from interested families. We will ask the doctor or hospital, or both, to send us all of your child’s medical records.
We will look at the records carefully to help us understand your child’s medical history and current situation. This information helps our multidisciplinary team develop an individualized plan for your child’s evaluation visit. For example, if your child has recently had certain laboratory or imaging tests done, we won’t repeat those tests unless there’s a valid medical reason for doing so. Understanding how the disease is affecting your child helps us identify which specialists your child should see during the evaluation.
It’s important for families to know that undergoing a pre-transplant evaluation involves no commitment on either side. It carries no guarantee that your child will be listed for a transplant or, conversely, any requirement that you must agree to have your child placed on the transplant waiting list. We can answer questions, provide information, and make recommendations. Ultimately, however, the decision to proceed with a transplant, or not, is a personal one for each family to make.
The evaluation is an opportunity for the family and the health care team to meet and get to know each other, as well as for the family to gather information and get answers to any and all questions you may have. We hope you’ll feel comfortable raising any concerns. Please don’t hesitate to ask us about any issue that’s on your mind. There are no dumb or silly questions. And, of course, if after you’ve gone home you think of something that you wish you had asked, please give us a call.
You can expect that the evaluation will be a two- or three-day event. The staff of our Center for Rare Disease Therapy will work with you to arrange for you, your child, and other family members to stay near the hospital, either at our Ronald McDonald house or at a nearby hotel, while you’re here for the evaluation.
We’ll send you a schedule in advance of your visit. This will tell you which medical and surgical specialists you’ll be seeing at what times and what laboratory or imaging tests we would like your child to have during the evaluation. To the extent possible, we try to anticipate all the testing we’ll need so that it’s a relatively smooth process while you’re here.
Please tell us more about what we can expect during our child’s evaluation.
Because PA is a genetic disease, the specialists you’ll see will likely include a medical geneticist and a metabolic dietician. Also, because PA often causes heart problems, your child’s evaluation is likely to include basic heart function tests and an assessment by a cardiologist. Depending on how the disease is affecting your child, the evaluation may also include visits with specialists such as the following:
- A neurologist, to assess brain function
- A gastroenterologist, to assess pancreas function
- A hematologist, to assess bone marrow function
Although we try to anticipate all the testing we’ll need and schedule it in advance, sometimes we may decide that it would be helpful to do an additional test that wasn’t originally on the schedule. For example, depending on the results of the basic heart function tests, the cardiologist might want to do a “stress test” that will provide more detailed information and measurements relating to how well your child’s heart is functioning.
If we decide to go ahead with listing our child for a transplant, what are our options for obtaining a donor liver? How long can we expect it to take to find a compatible donor?
PA is considered a high-priority condition for liver transplantation, so your child’s name will be near the top of the waiting list. However, because demand for donor livers is high and supply is limited, I tell families to be prepared to be on the waiting list for several months.
With any liver transplant, careful testing needs to be done to ensure compatibility of the donor liver and the transplant recipient.Many factors can influence the waiting time for a compatible organ. For example, a child with an uncommon blood type may face a longer wait.
In general, child-size donor livers are scarce. A unique feature of the liver, however, is that it is the only organ in the human body that can regrow. This means that in some cases it’s possible to transplant a section of a healthy liver rather than the whole organ. For example, a child who needs a liver transplant may receive a section of a liver from an adult donor. You may hear this type of transplant referred to as a “reduced-size” or “split” liver transplant.
Another type of liver transplant involves a living person – such as a relative, friend, or even a stranger – donating a section of their liver to someone who needs a transplant. Living-donor transplants may be an option for some children with PA. However, because PA is a genetic disease, parents and possibly siblings may be carriers of one of the genetic defects that cause the disease. Someone who is a carrier would not be a suitable living donor.
The good news is that children who receive a partial liver seem to do just as well as those who receive a whole liver. All of the options for obtaining a donor liver, including a reduced-size, split, or living-donor transplant, are discussed during the pre-transplant evaluation.
We’ve decided that a liver transplant is right for our child. What are the next steps?
When your child’s name is placed on the liver transplant waiting list, we will give you a pager that you will need to take with you everywhere you go so that we can reach you right away when we get a call that a matching donor liver is available. We don’t know when that call will come, but when it does you’ll need to be able to get to Children’s Hospital in a safe, but timely fashion. The transplant team will work with you to establish a ‘travel plan’ for you and your family for when the transplant is likely to occur.
While your child is on the waiting list, our specialists will work with your local doctors to care for your child and optimize their medical condition ahead of the transplant.
We know that waiting can be a difficult time for families. Your transplant coordinator is always available to respond to your questions and concerns and can also help you make travel arrangements.
Once you arrive at the hospital, preparations for the transplant may take from 12 to 24 hours. Your child will undergo another round of tests to confirm that the donor liver is a good match. Your child will also need to fast before surgery. Our metabolic dieticians will help us prepare intravenous fluids to provide your child with an individualized balance of fats, protein, and glucose to maintain stability while they can’t take anything by mouth.
The liver transplantation surgery may take up to several hours, although this varies in each case. While your child is in the operating room, a member of the transplant team will keep you informed on the progress of the transplant.
After the surgery, your child will go to the intensive care unit to be monitored closely until their condition is stable. Then your child will be moved to the liver transplant unit. Staff here will help you learn about your child’s medications, diet, need for follow-up care, and anything else you’ll need to know to care for your child.
After the transplant, will our child have to take anti-rejection medication?
After a liver transplant, you should expect that your child will need to take medication for the rest of his or her life to prevent organ rejection. The body’s normal reaction to a transplanted organ is to recognize it as a “foreign agent” and mount an immune response against the new liver. Anti-rejection medications suppress the immune system, which is the body’s defense system against illness and infection, to prevent it from attacking the new liver.
Because anti-rejection medications weaken the immune system, your child may be more likely to get infections – and those infections will be harder to treat. You will need to notify the transplant team at the first sign of an infection, such as a fever, chills, sweats, coughing, nasal congestion, diarrhea, redness or swelling, pain, or vomiting. A referral to a doctor may be needed as well.
With immune-suppressing medications, the goal is to find a treatment plan that achieves the needed degree of immune suppression while causing the fewest and least harmful side effects. Regular blood tests will help your child’s doctors monitor the medications’ effectiveness.
The risk of organ rejection declines over time. This means that in time your child should be able to take lower doses of anti-rejection medications. Most likely, however, he or she will need to continue taking at least a low dose of immune-suppressing medication lifelong.
Here at Children’s Hospital of Pittsburgh of UPMC and elsewhere, research is underway to learn more about whether some liver transplants patients can eventually stop taking immune-suppressing medication without increasing their risk for rejection of the transplanted organ. This research is a long-term effort, however, and it will be years before we can answer this question.
For more information, please visit: www.chp.edu/rarecare or call (412) 692-RARE (7273)
In Part 2 of this article, Dr. Squires will summarize the findings of a recent study of outcomes in children with PA and methylmalonic acidemia who received liver transplants at Children’s Hospital of Pittsburgh of UPMC.
 Fundraiser for Propionic Acidemia Foundation (PAF) in memory of Lauren and in honour of Jenna
Fundraiser for Propionic Acidemia Foundation (PAF) in memory of Lauren and in honour of Jenna transitioned to a program called Gateway To Adulthood (GTA). Jenna’s metabolic status has been stable. However, last year when Jenna turned 19 she suddenly had her first seizure. It was a scary time for us as we didn’t understand why she developed epilepsy. It was happening often. With a metabolic crisis, we knew our protocol. Yet, with seizures we had to be alert and constantly in Jenna’s presence, as it could happen at any time.
transitioned to a program called Gateway To Adulthood (GTA). Jenna’s metabolic status has been stable. However, last year when Jenna turned 19 she suddenly had her first seizure. It was a scary time for us as we didn’t understand why she developed epilepsy. It was happening often. With a metabolic crisis, we knew our protocol. Yet, with seizures we had to be alert and constantly in Jenna’s presence, as it could happen at any time.









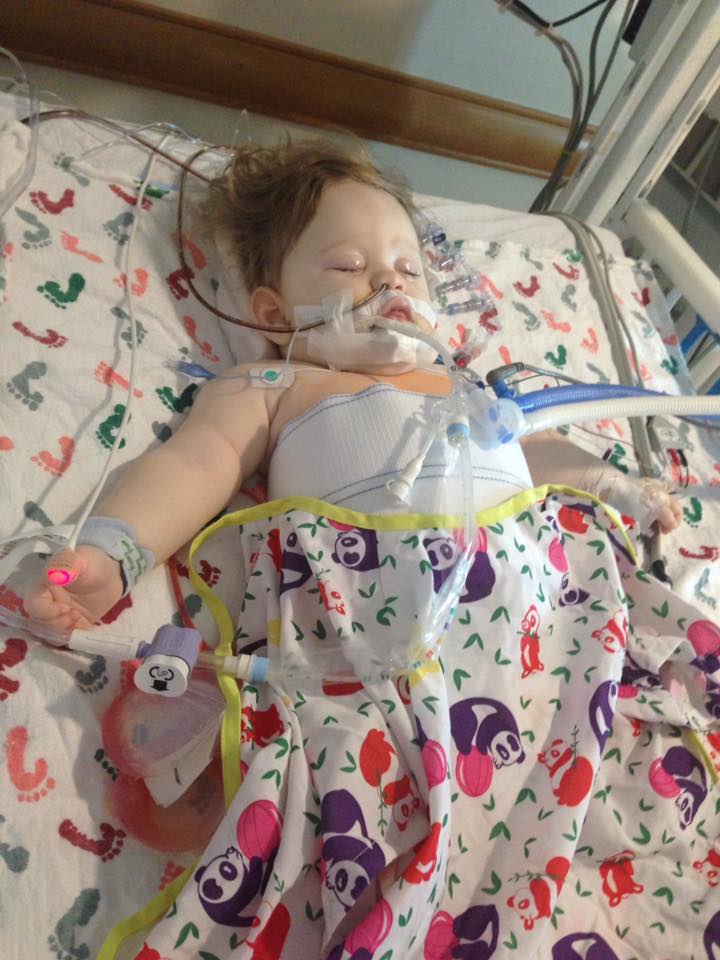
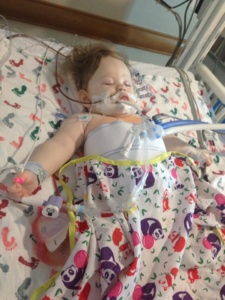 were very fortunate that our first call was “the call” that gave Annabelle her new liver. She went back for surgery around 10:30pm that night and they finished her surgery around 9am that next morning. After surgery Annabelle spent about one week in the PICU. After that week the transplant team moved herto the transplant recovery unit where she stayed until she was discharged. Around two weeks post-transplant Annabelle did encounter a small episode of rejection. Even though “rejection” sounds scary it is very common early on in transplant, and mild cases like Annabelle’s are generally treated with some high-powered IV steroids for a few days. Annabelle was discharged on August 30th and only spent a total of 21 days in the hospital. The transplant/genetics teams in Pittsburgh told us to prepare for complications (as is common with Organic Acidemia patients), but overall Annabelle had very few complications from her transplant surgery for which we are thankful.
were very fortunate that our first call was “the call” that gave Annabelle her new liver. She went back for surgery around 10:30pm that night and they finished her surgery around 9am that next morning. After surgery Annabelle spent about one week in the PICU. After that week the transplant team moved herto the transplant recovery unit where she stayed until she was discharged. Around two weeks post-transplant Annabelle did encounter a small episode of rejection. Even though “rejection” sounds scary it is very common early on in transplant, and mild cases like Annabelle’s are generally treated with some high-powered IV steroids for a few days. Annabelle was discharged on August 30th and only spent a total of 21 days in the hospital. The transplant/genetics teams in Pittsburgh told us to prepare for complications (as is common with Organic Acidemia patients), but overall Annabelle had very few complications from her transplant surgery for which we are thankful.

{kind=link}