|
Hi, our son Gabriel was born in London. He was diagnosed with propionic acidemia at 2 weeks of age after spending his first hours of life hyperventilating and with severe acidosis and high ammonia. Luckily, the medical team at Great Ormond Street Hospital for Children was very good at stabilizing him and at diagnosing him very quickly. Despite never showing actual fits, Gabriel was then diagnosed with infantile spasms (a type of childhood epilepsy) at 5 months of age after deteriorating progressively. We were again very lucky that he responded well to treatment with vigabatrin. He was weaned off medication at one year of age and has been seizure free since. At that same age, he stopped eating by mouth completely and a g-tube was inserted in his stomach for feeding. His formula currently comprises pediasure, polycose, XMTV1, vitamins and flax oil. He is been very stable metabolically for the past two years and is been followed up by the metabolic team at Children’s Hospital of Philadelphia every six months. Gabriel’s development was very slow until 18 months of age but he has made tremendous progress since he started therapies with the Early Intervention Program when we moved to the United States. He just transitioned to a special education pre-school program where he continues receiving OT, PT and ST. Since he started school, his gains have been really amazing. He just started talking and is able to comprehend perfectly and speak words in all three languages he hears. He has also gained cognitively and matured a lot. He has still a lot to catch up being his major challenges his low tone and his speech but we embrace his enthusiasm and effort and try to offer him as much support as we can. —————————————————————————- Updated 9/2015 – Gabriel – 13 anni As is often the case with so many children who are born with an organic acidemia, Gabriel’s first year of life was full of difficulties and hospital stays. After his initial crisis at 24 hours of age, Gabriel had lots of episodes of metabolic decompensation, and high ammonia levels, compounded by undiagnosed epilepsy (infantile spasms). Nevertheless, he started to stabilize around the time of his first birthday, and you can check out a summary of Gabriel’s first five years of life in a previous report we wrote for PAF.
During his first year of life, Gabriel spent 9 months with a nasogastric tube, which was so uncomfortable that it greatly contributed to his decision to stopping eating. But he got a G-tube at 15 months of age, and quality of life improved, not only for him, but also for the rest of the family. Through the G-tube, we started managing him much better at home because we did not have to rush him to the hospital every time he started throwing up. He became even more stable at the age of 3, and he started preschool also then. At that time, Gabriel didn’t have words and was incredibly delayed in all domains (especially motor and language) but he has overcome a lot and has been steadily progressing ever since. This was a little miracle to him thanks to an amazing teacher. Infatti, one very important thing for us is that Gabriel has not stopped developing, even though he makes progress at this own pace (meaning, very slow!).
The other little miracle we experienced with Gabriel is that at the age of 6 we found a wonderful feeding therapist (who was actually a special-ed teacher with experience in feeding issues from the behavioral point of view). She taught Gabriel how to eat (after 6 years of being 100% tube-fed). It was a very slow process but Gabriel now eats like 50% of his nutrition by mouth with a very wide variety of (very healthy!) foods: rice, pasta, vegetables, fruits and so on, everything with low protein, although he also eats the occasional egg or fish sticks. The reason why he only eats like 50% by mouth is because he still has very poor oral motor skills, and is very slow chewing and swallowing. Regardless, the fact that Gabriel can eat by mouth has helped tremendously to normalize our family: we go more often to restaurants, and Gabriel is happy to seat at the table with us and even request his own food from the menu. He has gained so much confidence as a result of eating by mouth!
In practical terms, the biggest challenge that Gabriel faces right now is his executive function (he has very poor coordination and motor skills, although he can run relatively well) and his language (he is diagnosed with a language disorder and although his IQ is a bit low, the psychologists think that part of the problem is his severe language issues). As a result of this, Gabriel attends a special-education school with highly individualized education. His class has two teachers and seven other children, and Gabriel still receives a lot of therapy at school (OT, PT, ST).
What is crucial about Gabriel’s life is that he is a very happy child. He loves water and has been learning how to swim for two years now and is able to execute quite a nice stroke (although the coordination with breathing is very difficult for him). In the past two winters he has also been trying adaptive ski and has absolutely embraced it. He has been traveling quite extensively around the world since he was 2 months old (we go often to Mexico and Spain to visit our families, and he loves playing with his cousin Marifer). Last year, the Make-a-Wish Foundation granted Gabriel a trip to Japan. He wanted to visit Tokyo, the city where Lightning McQueen (from the movie “Cars 2”) races in the middle of the night lights. It was an absolutely amazing experience and a very beautiful memory that hopefully will last forever in all of us!
Looking forward, we think Gabriel’s life will be very challenging, as he is unlikely to be able to live independently. Tuttavia, we feel blessed that he is such a kind and caring child who tries to enjoy new adventures, no matter the challenge. So, we try not to think too hard about what it will be, but focus instead on who Gabriel is and what he does right now, taking one day at a time!
Please, feel free to contact us if you would like to learn more details about Gabriel’s management or activities.
Cheers,
Marisa Cotrina and Juan Carlos López |
Author Archives: Angela Waits


 At the age of 5 we found a school that could target much better his language delays and lack of socialization skills. He spent 5 incredible years at this fantastic setting. It was a great program for him. Although still very delayed, Gabriel now chats a lot and understands and speaks both English and Spanish (both his father and mother are native Spanish speakers) although he is clearly dominant in English. He now has a few friends at school and shows more interest for group activities, like soccer. He is no longer afraid of noise at the movies and initiates conversations with other children in the park. Academically, Gabriel is quite delayed but the fact that he is reading and writing is a miracle to us. He is also able to do very simple math.
At the age of 5 we found a school that could target much better his language delays and lack of socialization skills. He spent 5 incredible years at this fantastic setting. It was a great program for him. Although still very delayed, Gabriel now chats a lot and understands and speaks both English and Spanish (both his father and mother are native Spanish speakers) although he is clearly dominant in English. He now has a few friends at school and shows more interest for group activities, like soccer. He is no longer afraid of noise at the movies and initiates conversations with other children in the park. Academically, Gabriel is quite delayed but the fact that he is reading and writing is a miracle to us. He is also able to do very simple math.
 For the past year and a half, Gabriel has been learning how to cook with a special-ed teacher. Although Gabriel is not very big eater he enjoys cooking very much, and we decided to develop a program whereby he could learn new skills and gain independence at the same time. So, every Saturday he prepares a shopping list for a new recipe, goes to the supermarket, chooses the ingredients and pays. Ideally, we are aiming for him to do all this independently at some point. When he gets home, he starts smelling, chopping and stirring. He is now able to turn the gas knobs on, boil water and add salt and pepper to a simple dish! He really, really enjoys cooking, and this is the one thing that takes him away from his video games and such. His new addition to the menu has been “pumpkin pie cheese cake”. He just cannot have enough of it! But he cooks all kinds of dishes from a Thai salad to Mexican corn soup. Although he often does not like the new dishes he makes, he always, always tries them. This plan has been really working very well to the point that Gabriel has expanded the repertoire of foods and flavors he eats now, he is no longer afraid of trying new foods away from home, and he is slowly gaining some skills that, we feel, will be valuable in the near future.
For the past year and a half, Gabriel has been learning how to cook with a special-ed teacher. Although Gabriel is not very big eater he enjoys cooking very much, and we decided to develop a program whereby he could learn new skills and gain independence at the same time. So, every Saturday he prepares a shopping list for a new recipe, goes to the supermarket, chooses the ingredients and pays. Ideally, we are aiming for him to do all this independently at some point. When he gets home, he starts smelling, chopping and stirring. He is now able to turn the gas knobs on, boil water and add salt and pepper to a simple dish! He really, really enjoys cooking, and this is the one thing that takes him away from his video games and such. His new addition to the menu has been “pumpkin pie cheese cake”. He just cannot have enough of it! But he cooks all kinds of dishes from a Thai salad to Mexican corn soup. Although he often does not like the new dishes he makes, he always, always tries them. This plan has been really working very well to the point that Gabriel has expanded the repertoire of foods and flavors he eats now, he is no longer afraid of trying new foods away from home, and he is slowly gaining some skills that, we feel, will be valuable in the near future.Lucy H.
Lucy H. – updated Fall 2016
Il mio nome è Lucy e ho PA. I am 18 years old and graduated High School in May. I also have: Autism, Aspergers, Prolong QT Syndrome, and some intellectual disability. All of these things make some parts of my life difficult for me to handle. A volte, quando devo passare attraverso certe cose come i miei problemi di salute si può ottenere un po 'stressante per me. Tuttavia Sono così felice che, anche se io sono l'unica persona nella mia famiglia che ha bisogno di speciale che quindi in assoluto, la mia famiglia e tutti i miei cugini mi amano molto in ogni modo e amano avere intorno a me. Ora voglio raccontarvi un po 'della mia famiglia. Besides my parents I have: two siblings, a dog and two cats. Out of everything I do for Physical activity my favorite would definitely have to be Ballet Dancing. I started doing Ballet at Nel mese di luglio 2015 my mom and I traveled with my cheer team to Los Angeles for the Special Olympics world games. We cheered for athletes at lots of the sports events and we got to perform our routines. We became celebrities, we were on the news, people stopped us everywhere for pictures and autographs. It felt really cool to be famous. I also did Rhythmic Gymnastics for a couple years for Special Olympics as well, e ho vinto un sacco di metalli oro. Ho anche fare ha bisogno di una speciale concorso di bellezza, ogni anno nel mese di aprile. In 2014 I won the whole pageant and I got crowned as: Young Ms. Fabulous. I got to ride on a float in the Christmas parade downtown that year in December. I was freezing!!!!! However I thought that was really super cool.!, because I had never done anything like that before. I have a collection of 24 American girl dolls. I still play with them constantly when I am not at school, and its my favorite hobby. Quando gioco con loro allevia un sacco di mio stress. In middle school people made fun of me for playing with them at my age, but now I just over look those people, because its what makes me happy. My favorite thing to do with them is brush out and style their hair. Especially with braids. As far as school goes I am really good at math. I have taken in high school Algebra and Geometry. I was definitely better at algebra. I was in a resource Special Ed. English class, because I can’t comprehend that well when I read. I can read very well, but I can still only comprehend at about a 3rd or 4th grade level, so I choose to often read those books so that I can actually enjoy the story. When I was a Junior I went to my school prom for the first time. I took my friend Niki to the prom with me because I didn’t want to go with a guy. When I went to prom it was not like I had imagined it being, but I thought it was so cool and I was overly excited to be going!!!! I danced that whole night constantly. But by 11:00 pm I was ready to drop and I fell fast asleep on the way home. Some children & teenagers who have a disability don’t have very many friends or any at all. I consider myself lucky because I have a whole lot of friends. And they have all been also very supportive of me having a disability and this health condition. My Summary: The older I have gotten the more I have been able to do with taking care of myself and my diet. I can now I graduated in May from high school. For my career I want to take care of as many children as I possibly can with all kinds of different special needs and disabilities. I want to help them learn and grow just like other normal children. To make this happen, I will be working with Vocational Rehab to explore future job possibilities and the further education I will need to do these jobs. To sum it all up I have to face a lot of challenges in my life especially with learning because of my cognitive delays and learning disability. I always try to put forth all the effort I’ve got in everything I do. And in general I am a very caring and compassionate person. Everyone in my family and at school tells me all the time you are an extremely good friend. I think they are right about that. Having a disability makes it so that unfortunately I am a little less mature in some ways than other people my age. But sometimes I feel like that is what makes me who I am and also what makes me special. A lot of people have told me throughout my life in high school that I am a really strong girl. I really do try to be strong even when I don’t feel that way, but sometimes I have no other choice. I am a very smart and determined teenager. I am always determined to do whatever makes me most happy in life. The motto I have for myself is: “ You can do anything you want to as long as you stay strong and put your mind to it. With faith all things that you want to happen in life are possible you just have to believe in yourself.” I plan to use this quote for the rest of my life. formula with supervision and I can do all of my own tube feedings. I only use my tube for my formula. The more we have learned about I graduated in May from high school. For my career I want to take care of as many children as I possibly can with all kinds of different special needs and disabilities. I want to help them learn and grow just like other normal children. To make this happen, I will be working with Vocational Rehab to explore future job possibilities and the further education I will need to do these jobs. To sum it all up I have to face a lot of challenges in my life especially with learning because of my cognitive delays and learning disability. I always try to put forth all the effort I’ve got in everything I do. And in general I am a very caring and compassionate person. Everyone in my family and at school tells me all the time you are an extremely good friend. I think they are right about that. Having a disability makes it so that unfortunately I am a little less mature in some ways than other people my age. But sometimes I feel like that is what makes me who I am and also what makes me special. A lot of people have told me throughout my life in high school that I am a really strong girl. I really do try to be strong even when I don’t feel that way, but sometimes I have no other choice. I am a very smart and determined teenager. I am always determined to do whatever makes me most happy in life. The motto I have for myself is: “ You can do anything you want to as long as you stay strong and put your mind to it. With faith all things that you want to happen in life are possible you just have to believe in yourself.” I plan to use this quote for the rest of my life. – Lucy
———————————————————————————————————————————- Fall 2013 Hello friends,My name is Lucy and there are a lot of things that I do to contribute to this world even though I have Propionic Acidemia. There are lots of new things I just started doing this year and it has been unbelievable . It has created a whole new world for me. I am 15 and a sophomore at HCHS in Lexington, KY. My favorite classes this year are child development and chorus. Last year my favorite class Algebra and I it was really hard for me but I still was able to get a b. This year I take biology, world civ, geometry and English. I have to have a teacher make sure I stay on task and sometimes write for me because I can’t write quick enough to keep up. We do a lot of homework and mom has to drink a lot of wine to get through it.I get help with making sure my tube feedings get done at school, I do them on my own but I still have an adult to make sure they get done on time. I take my lunch that mom and I decide on and measure at home. An adult makes sure I eat everything and if I eat something else, I call my mom to check and I bring home the package. At lunch you don’t have to sit with your specific class anymore and you can sit wherever you want. I like going to school because I get to see my friends everyday. There are some people that make fun of me and are never going to change. My fishy smell is a big issue. When people bully me I try to go to the principal or the counselor. They try to help but kids don?t always listen. Sometimes I get down in the dumps about this, but don’t want to talk about it because they will call me a snitch. This year the most exciting thing was starting special Olympics cheer leading. This has opened up a whole new world for me because I made a bunch of new friends. It is a very good sport to do with friends because we work as a team. We went to competitions in Atlanta and here in Lexing-ton. We have the coolest makeup and this year we got new uni-forms with lots of sparkles. We have one of the most well known special Olympics cheer teams and were undefeated for 11 years. We have lots of people who can do flips and handsprings. I can do a forward and roll, an OK toe touch jump and I am working on a cartwheel but am trouble with my arm strength and straight legs. Cheersport competition in Atlanta was amazing. We won second out of 13 teams in the Special Needs Elite Division and I got a Cheersport jacket because it was my first year. I was so excited about being up on that stage with my team. I told my mom it was the best day of my life. Even though I am 15 my favorite hobby is to play with my American girl dolls. I have 18. I also have been doing dance since I was 3. I go to a church dance program with my friend Anna, but this year I am going to stick to ballet and work with my elementary school dance teacher.It feels like I?ve been able to eat this well since birth now. I really love to eat. Some of the things I eat are chips, pretzels, crackers, goldfish, strawberries, pineapple, apple, grapes, bananas, blueberry, tomato, mushrooms, cooked onions, cooked cabbage, corn, carrots, broccoli, beans, peas, edemame, sweet potatoes, pizza, pasta, french fries and my dad and I go get Chi-nese once a week (veggies and rice for me). I also drink things like water, juice, milk, and sprite. I don’t drink my formula though because it tastes bad. This year I was also in a Special Needs pageant where I won second in the talent part. I sang a song from a Bar-bie movie about friends and I sang acapella. I have a good voice and I like to sing. I went to Therapeutic camp this summer. I didn’t get to do Riding for Hope with the horse park this year because we were too busy with cheer. I was taking a tumbling class until I had to have surgery on my umbilical hernia and I will start that back soon. I took a musical theatre class at the Lexington Children’s Theatre one week this summer. It was called “Show your Wicked Side and we sang lots of songs from Wizard of Oz, Wicked, and The Wiz. I sang a solo part in Easy on down. After that we went to Chicago the first of August for a Special Olympics Gymnastics camp. It was great but really wore me out. Mom has been worried about me ever since we got back because I have been taking lots of naps. Dad says I need more exercise and they are making me take lots of walks and bike rides. The best part was I got to go to the American Girl store and have dinner. I have a CLS worker who started in March. Her name is Miranda and she makes me do a lot of chores. We go to the grocery and she makes me estimate costs and choose which brand to get. I always get the cheapest. We exercise, cook, run errands, go to the library, do laundry, work on reading comprehension, go shopping at the mall and she makes me count out the money. We had a yard sale and I sold all my barbies so I could make money for Chicago. Miranda made me add up the sales and count out the change. Even though she makes me work I like her and we do fun things too like watch movies, have brunch with her niece, and get manicures. This sum-mer she helped me learn to wash my own hair. I figured out I could wear my goggles in the shower and the water wouldn’t get in my eyes. That is really great!I am back in school now and Cheer starts again next week. We are really busy with all this and mom says it wears her out, but it is fun! That’s about all I have to tell for now.Love –Lucy ps. I love to text and email so send me a message sometime. —————————————————————————- About Me: Written by Lucy
My name is Lucy Caroline H. I am eight years old. I am a little girl who goes to Cassidy School. I am in the second grade in Mrs. McCormick’s class. I can read level two books; my favorites are Wings on Things, Junie B. Jones, and Hopscotch Hill books. Even though I can read them very well I like them better when my mom or dad reads them to me. My best skills at school are recess, 100% A Spelling Tests, and I am getting better with having green behavior cards (that means no arguing and talking back).
I go to Mrs. Kimball’s dance class on Monday’s and Thursday’s after school. We learn how to do arabesques, kicks and sashays. Last week we were in the variety show at my school. We performed a dance to “It’s a Hard Knock Life” from Annie and all of us dressed up like orphans.
I have tube feedings but I like to eat too! I like French fries, corn off the cob, asparagus, spaghetti with butter and salt, and most of all I love, love, love to cut up mushrooms, cook them with dad and eat them up.
I went to Disney this January with Make-A-Wish. I got to dance with Minnie Mouse and the Princesses, ride The Tea Cups Carousel, Dumbo, the Magic Carpet and all the rides. It was a wonderful trip.
I have two cats, one named Isabella and one named Samantha. Samantha is a kitten and I got her for an early birthday present. She is fun to play with and I try to remember to feed her. I am looking forward to summer when I can go tubing and swimming in Grandaddy’s lake in South Carolina. I love that!
About Lucy: Written by Mom
What can I add about Lucy. I can think of many words to describe her: Determined, Talkative, Dramatic, Strong-Willed. She has so much energy she wears me out with her determination. I have to laugh sometimes when I remember thinking she might not talk. This girl can talk your head off, and has a vocabulary beyond her eight years. Lucy’s favorite thing to do is pretend. She plays dress up, school, tea party, baby dolls, dance party, performs, sings, plays dollhouse, groovy girls, fashion pollys, barbies, pooh characters. She is all girl!!!!!!
Lucy is maintaining normal cognitive skills at school. We work really hard for this and do lots of repetition to help her with homework, extra help at school, tutoring, and therapies. She has the most trouble with focus and things requiring this focus. So far with repetition she is able to learn and every day it amazes me what she picks up that we haven’t worked on at all. Socially she is a little immature. She has trouble interacting appropriately with her peers, but we are working on this with social groups at school, coaching from teachers and us, playdates, social stories, ecc. We have seen progress in this area too, slow sometimes, then leaps. I worry most that she won’t have friends because she can be so bossy or inappropriate with her peers. We just have to keep working on this. In a way I am glad she doesn’t care so much about what peers think, because they can be cruel sometimes, especially as she gets older. She really loves her dance class and performing with the group. This takes place as school and the teacher focuses a lot on team building and recognizing each others strengths. It is a great way for the girls to get to know others in the school, not just the kids in her class. Her school has been great. She has an aid to do tube feedings and make sure she eats snack and lunch. She also has some extra help with her work when she needs it. Many of the teachers know how to do her tube feedings too. I have been very happy with her care there. She goes to church school weekly and this year she has her first communion which she seems to be pretty excited about. Her Propionic Acidemia is in fairly good control as long as she is healthy. We maintain strict diet intake, tube feeding and oral meals. Lucy enjoys eating, which we work really hard for. It is fun to take her to a restaurant and watch her order her “French fries with extra salt and a glass of water with a lid”. She also likes spaghetti, spaghetti O’s, Saute’ mushrooms with garlic, tomatoes, asparagus, bananas, goldfish crackers, pretzels, garlic bread, waffles, butter sandwiches, the list goes on. We check her blood amino acids and urine organics about twice a year now, unless she is sick. Her growth is stable. She is a big girl: about 79 lbs, about 53 inches tall. Lucy takes Atenolol daily for her Prolonged QT Syndrome which was diagnosed in December 2004. She also has a leg length difference which effects her balance. We monitor that through Shriner’s orthapedics and she wears a lift on her left shoe to help. We are not quite sure what will happen with that as she grows, we just have to wait and see had bad the difference gets. Just like normal families with kids we are busy. School, play dates, homework, work for mom and dad, and the pa extras thrown in. Lucy and I battle a lot, the mommy – daughter thing at its best. Many days I feel like I am talking to a teenager already the way she argues with me over almost everything. I definitely need my glass of wine every evening to get me through homework time. I am not kidding!!!!! After all it is my job to take care of her and often that means being the bad guy, but I am a good mommy, whether she likes it or not. HA! |


Carson A.

Carson A. We adopted Carson at birth and we have been blessed ever since. He’s the most amazing little boy! Carson is so silly and makes us laugh all the time. Carson has a 16 year old sister and a 19 month old brother. He is a very happy little boy, is extremely active and he enjoys music a lot. It’s amazing how much rhythm he has at such a young age. We think he will probably be a famous musician some day.
Carson will be Three years old on June 15th 2006. He was diagnosed with PA at three weeks of age by Doctor Jose Abdenur at Children’s hospital of Orange in California. We live in Laguna Niguel, California. Carson is doing very well. He is about 9months delayed in speech and cognitive. Physically he is only a little delayed because of low muscle tone but seems to keep up pretty well. He stopped eating around 9 months old and has been eating strictly by G-Tube ever since. He will put some food in his mouth and suck on it but never really swallows anything. He likes primarily salty things. He drinks water from a bottle and we have just graduated to a sippy cup. He currently weighs 36 lbs and is 37 in. tall. His diet consists of 126 grms. Propimex-1, e 102 grms of Similac to a total volume of 30 oz. He receives 21 oz. by bolas feed during the day and the rest at night with the pump. His medications are Carnitine-6 mls 3 xs per day, sodium benzoate-4 grms per day, Flagil-1.6 mgs 2x per day, and Prilosec 3.5 mgs 2 x’s per day. Carson receives two hours per week of OT therapy and two hours a week of speech therapy. Carson has low muscle tone, but it is not severe. We see Dr. Abdenur every two months and we have all his Aminos and Carnitine and Amonia levels checked. We work with a wonderful dietician and we change his formula according to his body weight and his labs. We Love our Doctor and can’t imagine where we would be without his dedication and Love to Carson. We know that we are so lucky to have him.
We would love to communicate with anyone. Here is our e-mail.
Our E-Mail is [email protected] |
Grace-Marie
| Grace-Marie |
At twenty eight weeks I went into pre-term labor with Grace-Marie. I was placed on bed rest and all things seemed to be going good under the circumstances.  Thirty days in the hospital being monitored and watched all the way up till Grace was born. I thought once it was over all the waiting and preventions we took to keep her inside me to grow were about to pay off. I would have a healthy bouncing baby girl. It kept me going, the thought of her kept me stabile and focused while I was in the hospital away from the world. Thirty days in the hospital being monitored and watched all the way up till Grace was born. I thought once it was over all the waiting and preventions we took to keep her inside me to grow were about to pay off. I would have a healthy bouncing baby girl. It kept me going, the thought of her kept me stabile and focused while I was in the hospital away from the world.
Grace-Marie was born on November 14, 2007 a day before her dad’s birthday. It was truly amazing. Her dad and my oldest son were there to coach me on while bringing Grace-Marie into this world. She was born at 7:30 a.m. 5 lbs. 2 oz.. I was so happy to finally see her for the first time. That’s when it first started Grace’s heart beat was going really fast so they wanted to check her out really good before I could hold her. The morning turned to night and I still could not see Grace. Grace would not eat so they wanted to watch her in the NIC (Neonatal Intensive Care) over night. One day turned into seven in the NIC. Grace began to eat a little but was not very responsive to touch. She also began to not keep what she ate down, therefore she was becoming very lethargic. No one knew what was wrong the doctors had not a clue, they kept waiting for things to change running different test and so forth. By the ninth day Grace-Marie was transfered to a new hospital the UCLA NIC unit. I continued to pray as I listened to the doctors tell me she would not move and they needed to get her somewhere, where the doctors knew more about helping Grace. Grace was not breathing on her own by this time and she stopped holding my hand. Once at UCLA the doctors just wanted to stabilize Grace. They wanted to track down her new born screening test with the hopes of finding information to formulate a treatment plan. The trouble with that was it was a Saturday and most places were closed. Grace’s dad and I were lucky that we had a doctor on staff that could get in touch with someone to get the results. Grace condition was really bad and they were not sure if she was going to make it. Once they received the results cretin things began to get ruled out. The doctor met with us and explained to us that Grace had a rare metabolic disorder and they were trying to narrow it between two that were in question. They wanted to start her on a dialysis right away to get the toxins out of her blood. The problem was they were not sure if it was going to work because she was so small. They took us into Grace and explained everything they were about to do. They had to place a small catheter in the side of her tiny neck. When I saw her laying there I felt so helpless and hopeless. I pulled together all the strength I had and decided that if this was the road I was dealt, I was going to have to take it and be strong for Grace. Propionic Acidemia is what Grace was diagnosed with. The two and half years of Grace’s life has been full of doctor appointments and emergency room visits. Some that have left me devastated. And other time the visits have adverted a crisis. These visit have given her a boost of energy so that she remains stable. Most of the time I spend praying and educating myself on the disorder. Grace was so dehydrated one time that they could not find a vain anywhere. They had to go into a bone in her leg. Yes, they had to drill a hole in her leg to access the point they needed. Well it did not go so well the first drill did not work so they had to try again on her other leg. Three long hours to get an IV started. Grace was beginning to decompose more and more and slowly slipping away from us. Well we made it through with God’s Grace, that was a two week stay in the hospital. The next month we got a permanent port placed for Grace. Grace now gets her blood drawn through her port and also all of her IV fluids when she is unstable. Grace is on a special diet to help her remain stable and receive all of the nutrients needed to grow. Then she all of a sudden did not want to eat anymore or drink anything. The doctors decided that a G-tube was needed to help her to remain stable and to continue to grow. We still have doctor visits once a month to flush her port and every three months to discuss her growth and diet with the genetics doctor. The common cold drives us crazy and keep us on our toes. Grace has physical therapy, speech therapy, occupational therapy, infant stimulation, and feeding therapy threw out the week which keeps me pretty busy. Sometimes it’s really hard. Most of the time I try to be thankful for every moment with her. Grace is one of the greats joys in my life. ——————————————– updated 8/28/18 Grace-Marie was born on November 14, 2007 a day before her dad’s birthday. She was born at 7:30 a.m. 5
La grazia è una delle grandi gioie della nostra vita,en |


Logan L
| Logan L |
 When my ex-husband called to tell me our son, Logan, was being transported by helicopter to Children’s Hospital of Wisconsin in Milwaukee I was in disbelief. Logan was a normal, healthy, active 15 year old. He was a sophomore in high school, eager to get his driver’s license and training for the school tennis team. Yes, he had been sick for a couple weeks with what seemed to be an upper respiratory infection and then possibly even asthma, but when I heard he had cardiomyopathy, I couldn’t believe it. I knew all too well this was an enlarged heart. When my ex-husband called to tell me our son, Logan, was being transported by helicopter to Children’s Hospital of Wisconsin in Milwaukee I was in disbelief. Logan was a normal, healthy, active 15 year old. He was a sophomore in high school, eager to get his driver’s license and training for the school tennis team. Yes, he had been sick for a couple weeks with what seemed to be an upper respiratory infection and then possibly even asthma, but when I heard he had cardiomyopathy, I couldn’t believe it. I knew all too well this was an enlarged heart.
Two years later Latreace was born, and then three years after that we had Logan. Both were healthy and routine wellness visits to their pediatrician never indicated there was any cause for concern. This was not supposed to happen again! Why? Within the next couple weeks of April 2005 we were question by doctors trying to figure out what the connection was between Justine’s and Logan’s heart condition. We also found medical advancements had made treatment much different. Within two days, Logan was listed for a heart transplant. But his condition was deteriorating quickly and he needed to have surgery to put him on a left ventricular assist device (LVAD) to support him while he waiting for transplant. Surgery went fine, but over the next few days we realized something was wrong. Logan was in status with subclinical seizures and not waking up. He had no previous history of seizures. Now neurologists were consulted. They could not control his seizures with standard mediation techniques and as an only option decided to try putting him in a deep drug induced coma. They started sending blood, muscle, skin and spinal fluid specimens for testing to laboratories throughout the country. Our daughter, Latreace, was also tested. She was fine. Some of the initial results pointed to the direction of some type of metabolic disorder, but we were told it would take months to know exactly what we were dealing with. In the mean time, while Logan was still in a coma, genetics was called in. They recommended starting Logan on a “cocktail” of supplements that would be beneficial to someone with his suspected range of disorders. Finally after a month, something worked. Logan was awake without seizures. Now he required intense physical therapy to get his body strong enough to be re-activated for transplant. June 18, 2005 the day came. This surgery went remarkably well and a week later we received the lab results we had been waiting for: Acidemia propionico (PA). Logan had an elevated C3 level in an acylcarnitine profile, and his urine tests showed chemicals that suggested it was PA. I had no idea what this was, but spent several weeks searching for information. Logan just didn’t seem to fit. He had never been sick as a baby or child like these other unfortunate children I was learning about. I was told Logan has a mild (insorgenza tardiva) case. I felt I should be thankful, but was rather puzzled by what a “mild” case was capable of. Skin and heart muscle were sent away for enzyme analysis for propionyl CoA carboxylase that showed he has 4% enzyme activity. While his enzyme activity is low, most children that are sick as newborns have 0% activity, which is likely why he is “mild” and didn’t have any symptoms until his cardiomyopathy appeared as a teenager. Logan left the hospital a month after his transplant and with the help of tutors was able to catch up on his studies and complete high school with his class. In many ways Logan has been a normal teenager. He does have many doctor appointments with cardiology, neurology and genetics and takes many medications and supplements, but he had been doing well. He has been attending Tech school and shared an apartment with several roommates. Unfortunately, gennaio 30, 2009 Logan was admitted to the hospital again. He is now In the last several years our family’s DNA mutation have been tested. It was confirmed that Logan’s father and I are both carriers of PA, and our daughter Latreace is also a carrier. I am sure there are other cases like Logan’s and I often wonder if Logan’s condition could have been prevented if he was diagnosed and treated early in life. Newborn screening for propionic acidemia is now done in most states in the US, but it is unknown whether infants with mild PA will be detected and treated to prevent sudden cardiomyopathy. Special thanks to Amy White, MS, CGC for assistance in preparing Logan’s story, and thanks to the entire talented and dedicated staff at Children’s Hospital of Wisconsin, Milwaukee. Debra L Logan (19) PA
On July 20, 2009, Logan received a new heart. On August 27, 2009, Logan chose to be in a better place. His battle was long but his love for life will always persist. Logan is an inspiration to us all. |
 Our oldest daughter, Justine, also a healthy child had been diagnosed with the same condition 20 years earlier when she was six years old. At that time the doctors told us they believed a virus attacked her heart. They could find no other cause and said this sometimes happens. There was nothing they could do for her. The subject of transplant was brought up, but quickly dismissed. Medical technology didn’t make this a realistic option 20 years ago. Within seven months she died. We had no other children at the time, but we were assured this should not be a concern for any future children we may have.
Our oldest daughter, Justine, also a healthy child had been diagnosed with the same condition 20 years earlier when she was six years old. At that time the doctors told us they believed a virus attacked her heart. They could find no other cause and said this sometimes happens. There was nothing they could do for her. The subject of transplant was brought up, but quickly dismissed. Medical technology didn’t make this a realistic option 20 years ago. Within seven months she died. We had no other children at the time, but we were assured this should not be a concern for any future children we may have.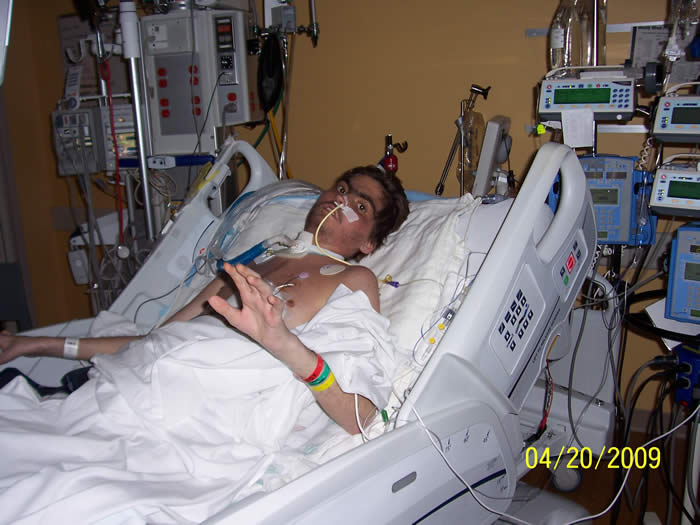 dealing with severe rejection and is in need of another heart transplant and possible kidney transplant due to poor profusion. He again is being supported by a ventricular assist device (BIVAD), dialysis and a tracheotomy. He is not healthy and strong enough to be listed for re-transplant yet, but we are hoping he will be soon. Genetics continues to be consulted to determine how his metabolic disorder plays a role in his symptoms and treatments.
dealing with severe rejection and is in need of another heart transplant and possible kidney transplant due to poor profusion. He again is being supported by a ventricular assist device (BIVAD), dialysis and a tracheotomy. He is not healthy and strong enough to be listed for re-transplant yet, but we are hoping he will be soon. Genetics continues to be consulted to determine how his metabolic disorder plays a role in his symptoms and treatments.
Angelica S
|
A challenging medical journey and a full loving life. Her faith, courage and strength are an inspiration. May her memory be eternal. October 20, 2008-Luglio 30, 2015
Our daughter Angelica had her first ‘metabolic crisis’ at three days Now she is a little over four months old, and while there have certainly been bumps in the road, it does not seem like we could have asked for a much better situation. She has an incredibly hearty appetite for her special formula (“power milk”) and is the sweetest, happiest, most easy going kid you could ever hope to see, and has even been letting her mom and dad sleep through the night, pretty much since we came home! She is tough (and stubborn) as well, having survived intensive dialysis at such a young age and showing no signs of damage from the ordeal.
Best Regards, Joe and Toula |

 old. Within twenty-four hours of bringing her home from the hospital after birth, she had become so lethargic and unresponsive that we had no choice but to contact the nurse on call, who instructed us to immediately call an ambulance. The staff in the emergency room (thankfully, we live less than ten minutes away from the fantastic resources of the University of Michigan Hospital) were able to stabilize her to a degree, but were very troubled by her ammonia levels and other test results, though they did not know the cause at that time. A conclusive diagnosis of PA was reached by the following day, and after another harrowing four days in intensive care undergoing hemodialysis she was moved to a moderate care wing. We were able to return home about a week after that, once she had regained enough strength, and more importantly, her appetite.
old. Within twenty-four hours of bringing her home from the hospital after birth, she had become so lethargic and unresponsive that we had no choice but to contact the nurse on call, who instructed us to immediately call an ambulance. The staff in the emergency room (thankfully, we live less than ten minutes away from the fantastic resources of the University of Michigan Hospital) were able to stabilize her to a degree, but were very troubled by her ammonia levels and other test results, though they did not know the cause at that time. A conclusive diagnosis of PA was reached by the following day, and after another harrowing four days in intensive care undergoing hemodialysis she was moved to a moderate care wing. We were able to return home about a week after that, once she had regained enough strength, and more importantly, her appetite. The pediatric genetics staff at the UM Mott Children’s Hospital are a great resource, and having them so close at hand is an incredible luxury that we definitely don’t take for granted. We are also blessed with a very competent and understanding daycare provider–they take everything in stride, whether it is diluting formula as needed, providing medications, or even ketone testing. With everything we have at our disposal, the inevitable problems we are going to encounter do not seem nearly as daunting as they once did.
The pediatric genetics staff at the UM Mott Children’s Hospital are a great resource, and having them so close at hand is an incredible luxury that we definitely don’t take for granted. We are also blessed with a very competent and understanding daycare provider–they take everything in stride, whether it is diluting formula as needed, providing medications, or even ketone testing. With everything we have at our disposal, the inevitable problems we are going to encounter do not seem nearly as daunting as they once did.Nicholas P
| Nicholas P. |
|
Luglio 24, 2001 – aprile 23, 2012 Nicks buoyant tenacity taught his family and friends how to love selflessly and carry on through hardships. His example will be a guide and comfort to those grieving his loss. He was a devoted aquarium enthusiast and played baseball with the Rivermont Dream League. He loved the River Park, his collection of movies and books (especially Cars), horses and his dog Buster.
Click below to sign his guest book and view more photos of Nick. |


Cindy X
| Family Story – Cindy X. |
 Hi, everybody. My name is Tad. Firstly I should say I am fortunate. I am fortunate to get the opportunity to join the university, where I met my girlfriend, who became my wife 10 years later. I am fortunate to get my present job as a petroleum geologist after I got my PHD degree from RIPED, where I met a lot of good friends. I am fortunate to have a beautiful daughter, Cindy, in 2012. Tuttavia, things changed from the day I received a call, which told me the results of my daughter’s lab test. The doctors suspected my 20-day old daughter to have PA. Looking at the cute baby, we all doubted the results. But we went to a doctor the next day, and did some normal lab tests. According to the tests result, the doctor didn’t think Cindy has PA. Then we went back home, with a burden unloaded. Hi, everybody. My name is Tad. Firstly I should say I am fortunate. I am fortunate to get the opportunity to join the university, where I met my girlfriend, who became my wife 10 years later. I am fortunate to get my present job as a petroleum geologist after I got my PHD degree from RIPED, where I met a lot of good friends. I am fortunate to have a beautiful daughter, Cindy, in 2012. Tuttavia, things changed from the day I received a call, which told me the results of my daughter’s lab test. The doctors suspected my 20-day old daughter to have PA. Looking at the cute baby, we all doubted the results. But we went to a doctor the next day, and did some normal lab tests. According to the tests result, the doctor didn’t think Cindy has PA. Then we went back home, with a burden unloaded.
But after that day, my wife got nightmares nearly every night. I have to say, I began to believe the saying of mother-daughter-one-heart. After one week, we decided to do some more professional tests to confirm the situation. About one months later, all the results reflected that Cindy do have PA, which is PCCB in subtype. The result shocked us, and even the doctor. She said she never see a PA baby in such a good condition during her career. Then we began to give her special diet with XMTVI Analog formula and SigmaTau L-carnitine. In the beginning we know little about PA, so we cut off every milk including her mother’s milk. What a pity baby and stupid parent!
I want to say I am fortunate again, thanks the God. We then met a professional doctor who told us how to feed Cindy. Before that we suffered a lot, even Cindy had to be sent to PICU once when she was 1year old. After that, Cindy began to sit, walk, talk at normal age. But then, things became worse, the XMTVI Analog was forbidden to sale on Mainland China. We have to change into another formula, OA1, which is difficult to buy in China. During the period, I joined in to an PA organization in China. We talked a lot there and learned a lot from each other. There are totally about 134 parents with PA or MMAW kids. The organization is really awesome! And as you all know, as most of PA kid, now Cindy begins to dislike the formula and her food, she wants to eat meat. She is now still in the hospital for the past 20 days eating less. So I want to change her diet according to my self’s knowledge, for we cannot get more suggestions from our doctors any more. I will add more formula with little protein but high energy and other minerals.
Now I am in the USA to do a co-research program leaving my family in China. One of the reasons why I choose to come here from such a long way and in such a critical time for my family is that I want to learn something about PA here in the USA |
Allison E
Allison E.
updated 4/2022 (as seen in the Spring 2022 Notiziario) 
Allison (PA), turned 16 in November. She is doing well. We recently stayed at Give Kids The World and visited several theme parks in Orlando. Allison's Make-A-Wish was to meet Belle. Originally she was to go in March of 2020 to dress like Belle and have lunch with her in the castle. Then came COVID...Fortunately we got to reschedule the trip. With all of us being vaccinated, we were able to go. Characters aren't meeting the way they used to, but Allison did get to see Beauty and the Beast live (front row!) and attend Beauty and the Beast sing-a-long. She saw many princesses and characters, including Cinderella in the Castle. She enjoyed rides, and firework shows. We did the Magic Kingdom, Epcot, Disney Springs, Hollywood, & Universal Studios. It was a very busy 4 days! The 5 nights we stayed at GKTW Village were amazing. So much to do, a Carousel, magic tree house (where the wish kids make a star that the fairy adds to the night sky), great food, ice cream, activities, gifts every day...even a spa! Allison got her hair done (pink), nails (polka dots) and even glitter tattoos. We were treated like royalty and it was just an amazing trip.
 We are back to reality & ready to schedule many follow up appointments with not only genetics, but orthopedic surgeon for her scoliosis, ophthalmologist for optic atrophy, dermatologist, dentist, etc...
We are back to reality & ready to schedule many follow up appointments with not only genetics, but orthopedic surgeon for her scoliosis, ophthalmologist for optic atrophy, dermatologist, dentist, etc...Michelle, Allison's mom
3/2015 
I can't remember the last time I wrote an article or update about my amazing girl...I also can't remember the last time she was in the hospital before now. It is Friday, March 20th, and she was admitted the evening of the 17th. Thankfully, hospital visits are less frequent the first few years of her life. We are here now because she has a stomach bug like the rest of the family, but couldn't get better on her own like we did. So...IV fluids (D10), Carbaglu, e "sick day" formula...
Nationwide Children's Hospital is an incredible place, with an amazing staff. Before I go on about Allison, I would like to thank some of the most special people in her life. "Daddy" Dave, who does EVERYTHING! Grandma Char, who cares for her and/or her twin brother Austin for countless hours whenever needed. Yvette Williams, her home nurse for over half her life, and full-time nurse at Nationwide Children's Hospital for 20 years this August! Dr. Bartholomew, Jimia Hoy, and everyone in the Genetics department, and all of the wonderful nurses on the 11th floor (many who have cared for Allison since she was a week old!) We love you all and couldn't do this without you!
Allison is currently in 3rd grade and is in a special needs class (another special person...Kelly Duell, intervention specialist who has taught her SO much, every year since kindergarten!) Despite her developmental delays, she is reading and writing many words, doing math on a calculator, and expressing herself more than ever! She wears DAFOs, receives physical, occupational and speech therapy weekly. She loves music (Frozen soundtrack, Laurie Berkner, and nursery rhymes, mostly), dancing, playing hide and seek, arts and crafts, swimming, and playing with her friend Gwen. There isn't much she doesn't like, all I can think of is storms, dogs (but she is getting better), and she isn't very interested in food. Primarily G-tube fed her special formula (which consists of Propimex 2, whole milk, MCT oil, water, and levocarnitine) she is only allowed 3-4 grams of protein a day by mouth. She will snack on her favorite..."white cookies" (mini vanilla Oreos introduced by Grandma Char), and occasionally have a little applesauce, juice, or taste of something (but when asked usually replies "no, I don't like that anymore").
Potty training has been one of our biggest challenges. We have tried lots of things including the potty watch, and just not using pull-ups, but underwear instead. Very messy! The combination of her diet, delays, and ulcerative colitis make it necessary for us to use chucks on her bed every night as well. I just paused and wandered off thinking about whether or not she gets teased or made fun of at school. I am back now because I don't want to think about that. I know I wasn't always a nice kid, especially when I thought someone wasn't "normal".
Our goal for Allison is to be as "normal" as possible. We would love to see her become independent, have a job she enjoys, and maybe even have a family of her own someday. Whatever she wants is our wish, and we have to help her discover what that is.
Michelle
Columbus, Ohio
P.S. (Just for fun)...I used to joke that Allison's "Make-A-Wish" was to meet Mick Jagger and the Rolling Stones. I even got her to say it a few times 🙂 We haven't applied for anything like this for her, because we really aren't sure what she would pick. I just asked asked her what she most wanted, what she wished for...she answered "I wish for Allison". I just love her! I wish for a cure for Propionic Acidemia and will continue to do my part fundraising and supporting the wonderful PA Foundation.
I wish to meet Mick Jagger, so if anyone reading this has any connections...
LOL
---------------------------------------------------------------------------------------
Allison Lynne was born on November 29, 2005 in Columbus, Ohio. A healthy little girl, weighing in at 6’3 oz with her twin brother Austin at 5’10oz, the future looked bright. It was a healthy normal pregnancy. On Allison’s 1 week birthday, we got a phone call from the pediatrician’s office that said her newborn screening was abnormal. The few days prior, we did notice that she had been very sleepy and difficult to feed and we had a Dr. visit scheduled for later in the week. But, when we Dr. asked us to bring her in to get more blood work done as well as a urine sample, I was feeling nervous.
weighing in at 6’3 oz with her twin brother Austin at 5’10oz, the future looked bright. It was a healthy normal pregnancy. On Allison’s 1 week birthday, we got a phone call from the pediatrician’s office that said her newborn screening was abnormal. The few days prior, we did notice that she had been very sleepy and difficult to feed and we had a Dr. visit scheduled for later in the week. But, when we Dr. asked us to bring her in to get more blood work done as well as a urine sample, I was feeling nervous.
After the tests, they sent us home to await the results. On Wednesday, we called and told them that she wasn’t eating and they said to bring her into the office immediately, and once they saw her, they sent us to Children’s Hospital—this was Wednesday night. That night she had many firsts: including a spinal tap, IV, Oxygen, and was placed into a warmer and spent the night in the PICU. (Pediatric Intensive Care Unit). On Thursday December 8th, she was diagnosed with Propionic Acidemia. She spent her first week in the hospital and came home with a new diet of Propimex 1, Similac, Biotin, and L-carnitine. Allison did well until her next hospital stay 4 ½ months later in May. Her acid reflux was the reason for this visit. After receiving Prevacid and Reglan she was back home. She still was not eating really well and in October she got a feeding tube to assist her in getting all of her nutrients.
Later- her white blood count showed up low and after consulting with an immunologist, she was diagnosed with an IGG deficiency. She received infusions in August, September, and October. Her blood was checked in December and her IGG levels came up and consequently has not had an infusion since. Allison’s development has been supplemented with Physical and Occupational Therapy and focusing on her Gross Motor Development. Allison was saying a few words, clapping her hands, and getting very close to walking, when she developed a virus. This sent her back to the Emergency Room on Monday January 15th, 2007, with symptoms of vomiting and showing Ketones in her urine. After a day of testing, including a MRI, Spinal Tap, C-Scan, EEG, and Chest X-ray and another night in the ICU, she was diagnosed with a virus. They think a virus caused metabolic crisis (due to her Propionic Acidemia), and resulted in swelling of the brain. A neurologist said she had “chorea” which was “secondary to swelling of the basil ganglia.” Her EEG showed decreased brain activity and her MRI showed the swelling or “lesions” as the neurologist referred to them. She is home now, but not herself (not taking food or bottle, not sitting up, not holding toys, not talking…) They told us it could take weeks or months to tell if damage is permanent, but that the swelling and chorea should go away.
Evan,en
Evan M. – updated March 2015
 Evan is now 8 anni. He had a partial liver transplant in March 2012. Whilst the 1st years was very tough, unfortunately he was diagnosed 6 months after transplant with Lymphproliferative Disease. A type of Lymphoma brought on by the immunosuppressants needed to stop his body from rejecting his new liver. He needed a few months of chemotherapy but thankfully got the all clear from this a few months later. Since this however, Evan has come along so so much.
Evan is now 8 anni. He had a partial liver transplant in March 2012. Whilst the 1st years was very tough, unfortunately he was diagnosed 6 months after transplant with Lymphproliferative Disease. A type of Lymphoma brought on by the immunosuppressants needed to stop his body from rejecting his new liver. He needed a few months of chemotherapy but thankfully got the all clear from this a few months later. Since this however, Evan has come along so so much.
The transplant wasnt a ‘cure’ but is a huge help in managing Evan’s condition and giving the best chance at living a normal life. He can tolerate alot more protein which in turn helps his development and generally makes him look healthier. He is in his 2nd year in the local special needs school and he absolutely loves it. He runs out to the bus in the morning and always has a smile on his face wen he gets off the bus when he gets home! He loves the attention and being with others his own age. His speech and communication is coming along although we still can’t converse with him and his eating is steadily improving. Although foods still have to be pureed, he is open to trying new things and experimenting and playing with food whereas before he showed absolutely no interest in food and mealtimes.
 He is still tube fed most of the time via a mic-key button in his stomach. His feed consists of Energivits, Nutrini and MMA/PA Gel and all medications we give to him via this tube. So even though he still picks up every bug and infection going and does have long bouts out of school and the odd admission to our local hospital the future is looking bright for Evan. There is no doubt life is tough living with PA, we do our best by our children.
He is still tube fed most of the time via a mic-key button in his stomach. His feed consists of Energivits, Nutrini and MMA/PA Gel and all medications we give to him via this tube. So even though he still picks up every bug and infection going and does have long bouts out of school and the odd admission to our local hospital the future is looking bright for Evan. There is no doubt life is tough living with PA, we do our best by our children.
Not everyone agrees with our decision to go for a Liver Transplant but even though the 1st year was tough we don’t regret our decision. Our thinking was that if something happened during or after the transplant at least we are doing everything possible and in our power to give our child the best chance possible to have a normal life. Life with PA is so unpredictable.
Sarah (Evans mam)

———————————————————–
Evan is 4 and he was diagnosed with Propionic Acidemia when he was four days old. normal pregnancy, normal delivery and evan arrived a healthy 7lbs 11ounces three days early. On day 3 he was getting very lethargic, he wasnt feeding and was breathing funny from his nose, like he had a cold. He was brought to the Special Care Unit by a nurse and we were told he would be back up soon so we presumed it was just to be checked out. An hour later, he was on a ventilator as doctors were afraid he would stop breathing. A number of tests were done and doctors in Cork narrowed it down to a Metabolic Disorder but they were not the experts in this field, The Metabolic Center of Ireland is in Dublin so Evan was transferred up when he was 3 days old where they diagnosed him the day after. Evan was in the hospital for 3 months until well enough to return home. Since then he has been admitted to the local hospital a handful of times, for flus, bugs and infections but thankfully since birth he has had no major decompensation. He has a good enough appetite and drinks his XMTVI Maxamaid orally during the day and eats about 6 of his restricted 8 exchanges of protein orally, tuttavia, anything he does eat has to be liquidised with no lumps whatsoever. He also has a continued feed during the night through PEG Tube. We travel to his doctor in Dublin every 3 months for check ups, where they take blood and check his levels are normal, and we speak to dieticians etc. Evan is a wonderful, happy, sociable, and boisterous 4 year old , he struggles with speech and communication which can get him frustrated and he has regular speech therapy, physio and occupational therapy. Evan has been on a waiting list for an Auxiliary Liver Transplant for 7 months now.