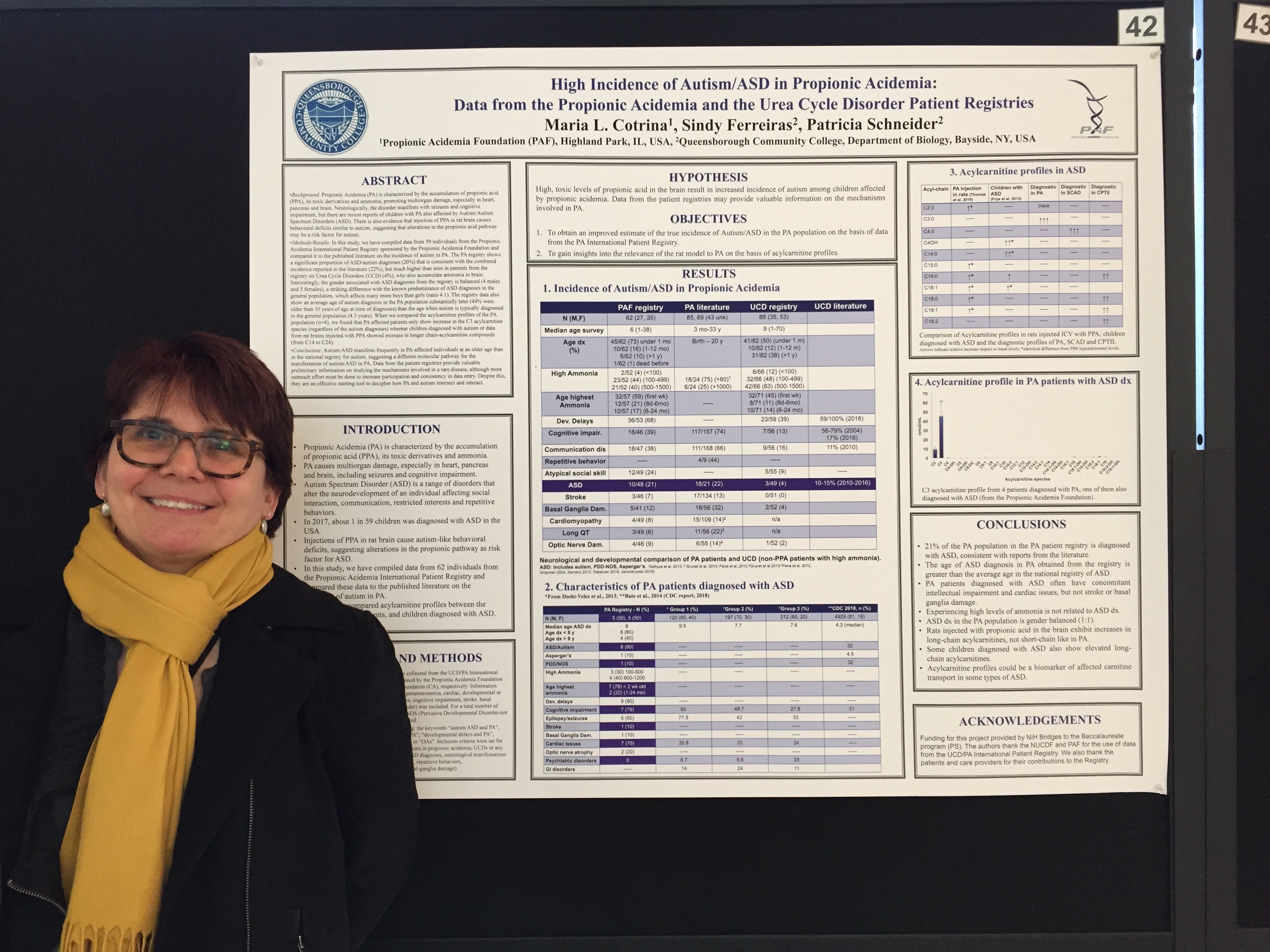
PAF Awards $33,082.12 Research Grant in 2019
PAF Awards $30,591 Continuation Grant in 2020

Eva Richard, PhD, Universidad Autonoma de Madrid, Spain
“Cardiomyocytes derived from induced pluripotent stem cells as a new model for therapy development in propionic acidemia”
Understanding the cellular and molecular mechanisms that occur in genetic diseases is essential for the investigation of new strategies for their prevention and treatment. In this context, induced pluripotent stem cells (iPSC) offer unprecedented opportunities for modeling human disease. One of the fundamental powers of iPSC technology lies in the competency of these cells to be directed to become any cell type in the body, thus allowing researchers to examine disease mechanisms and identify and test novel therapeutics in relevant cell types.
The main objective of this project is focused on the generation of human iPSC-derived cardiomyocytes (hiPSC-CMs) from propionic acidemia (PA) patients as a new human cellular model for the disease.In PA, cardiac symptoms, namely cardiac dysfunction and arrhythmias, have been recognized as progressive late-onset complications resulting in one of the major causes of disease mortality. Using hiPSC-CMs we will study cellular processes, such as mitochondrial function and oxidative stress which have been recognized as main contributors for PA pathophysiology. In addition, our aim is to unravel novel altered pathways using high-throughput techniques such as RNAseq and miRNA analysis. We will also examine the potential beneficial effects of an antioxidant and a mitochondrial biogenesis activator in PA cardiomyocytes. The results that derive from this project will be relevant for the disease providing insight into the affected biological processes, and thus providing tools and models for the identification of novel adjuvant treatments for PA.
Update April 2020 – Eva Richard PhD
Thanks to propionic acidemia (PA) foundation, we have developed a new cellular model of PA based on induced pluripotent stem cells (iPSC) with the goal of defining new PA pathomechanisms which could be potential therapeutical targets. Traditionally, disease pathophysiology has been studied in immortalized or human cell lines and in animal models. Unfortunately, immortalized cells often do not respond as primary cells and animal models do not exactly recapitulate patients‘ clinical symptoms. So far, patients-derived fibroblasts have been mainly used as cellular models in PA due to their availability and robustness, but they have important limitations. The ability to reprogram somatic cells to iPSCs has revolutionized the way of modeling human disease. To study rare diseases,
stem cell models carrying patient-specific mutations have become highly important as all cell types can be differentiated from iPSCs.
We have generated and characterized two iPSC lines from patients-derived fibroblasts with defects in the PCCA and PCCB genes; and an isogenic control in which the mutation of the PCCB patient was genetically corrected using CRISPR/Cas9 technology. These iPSC lines have been successfully differentiated into cardiomyocytes,
and their presence was easily established by visual observation of spontaneously contracting regions and by the expression of several cardiac markers. PCCA iPSC-derived cardiomyocytes exhibited reduced oxygen consumption, an accumulation of residual bodies and lipid droplets, and increased ribosomal biogenesis. Furthermore, we found increased protein levels of HERP, GRP78, GRP75, SIG-1R and MFN2 suggesting
endoplasmic reticulum stress and calcium perturbations in these cells. We also analysed a series of heart-enriched miRNAs previously found deregulated in heart tissue of a PA murine model and confirmed their altered expression.
The present study represents the first report of the characterization of cardiomyocytes derived from iPSCs generated by PA patients ́ fibroblasts reprogramming. Our results provide evidence that several pathomechanisms may have a relevant role in cardiac dysfunction, a common complication in PA disease. This new cellular PA model offers a powerful tool to unravel disease mechanism and, potentially, to enable drug
screening/drug testing. Despite improved therapy over the past few decades, the outcome of PA patients is still unsatisfactory, highlighting the requirement to evaluate new therapies aimed at preventing or alleviating the clinical symptoms. Additional research is required to determine the contribution of the mechanisms identified in this work to the cardiac phenotype and how this knowledge can help formulating better personalized therapeutic
strategies in the future.
We sincerely thank the Propionic Acidemia Foundation for supporting our investigation, which has resulted in a truly motivating experience for us, feeling we belong to the PA research family. The funding we received has led to important advances in PA pathophysiology, and our aim is to continue this research in the near future.
Update September 2019 – Eva Richard PhD
There is an unmet clinical need to develop effective therapies for propionic acidemia (PA). Advances in supportive treatment based on dietary restriction and carnitine supplementation have allowed patients to live beyond the neonatal period. However, the overall outcome remains poor in most patients, who suffer from numerous complications related to disease progression, among them cardiac alterations, a major cause of PA morbidity and mortality. In our research, we developed a new cellular model of PA based on induced pluripotent stem cells (iPSC) with the goal of defining new molecular pathways involved in the pathophysiology of PA which would be potential treatment targeting.
Traditionally, disease pathophysiology has been studied in immortalized or human cell lines and in animal models. Unfortunately, immortalizedcells often do not respond as primary cells and animal models do not exactly recapitulate patients‘ symptoms. So far, patients-derived fibroblasts have been mainly usedas cellular models in PAdue to theiravailability and robustness, but they have important limitations.
The ability to reprogram somatic cells to iPSCs has revolutionized the way of modeling human disease. To study rare diseases, stem cell models carrying patient-specific mutations have become highly important as all cell types can be differentiated from iPSCs. We have generated and characterized two iPSC lines from patients-derived fibroblasts with defects in PCCA and PCCB genes. These iPSC lines can be differentiated into cardiomyocytes that mimic the tissue-specific hallmarks of the disease. The presence of PA cardiomyocytes has been easily established by visual observation of spontaneously contracting regions, and the expression of several cardiac markers. We have observed that PCCA-deficient cardiomyocytes present an increase in degradation products and in lipid droplets, and exhibit mitochondrial dysfunction compared to control cells. We further discovered the down-regulation of several miRNAs in PCCA cardiomyocytes compared to control ones, and several miRNAs targets are currently being analyzed in order to investigate underlying cellular pathological mechanisms. Interestingly, we have performed several experiments to analyze the effect of the mitochondrial biogenesis activator, MIN-102 compound (PPAR agonist, derivative of pioglitazone) in cardiomyocytes.
Preliminary results showed an increase in the oxygen consumption rateof PCCA and control cells. In our next steps, we plan to complete the analysis in the PCCA cardiomyocyte line, characterize PCCB cardiomyocytes and to study in depth the therapeutic potential of MitoQ and MIN-102 compounds.
We would like to sincerely thank the Propionic Acidemia Foundation for supporting our research.
Update March 2020
“Cardiomyocytes derived from induced pluripotent stem cells as a new model for therapy development in propionic acidemia.”
Eva Richard, Associate Professor
There is an unmet clinical need to develop effective therapies for propionic acidemia (PA). Advances in supportive treatment based on dietary restriction and carnitine supplementation have allowed patients to live beyond the neonatal period. However, the overall outcome remains poor in most patients, who suffer from numerous complications related to disease progression, among them cardiac alterations, a major cause of PA morbidity and mortality. In our research, we developed a new cellular model of PA based on induced pluripotent stem cells (iPSC) with the goal of defining new molecular pathways involved in the pathophysiology of PA which could be potential therapeutical targets.
Traditionally, disease pathophysiology has been studied in immortalized or human cell lines and in animal models. Unfortunately, immortalized cells often do not respond as primary cells and animal models do not exactly recapitulate patients‘ symptoms. So far, patients-derived fibroblasts have been mainly used as cellular models in PA due to their availability and robustness, but they have important limitations.
The ability to reprogram somatic cells to iPSCs has revolutionized the way of modeling human disease. To study rare diseases, stem cell models carrying patient-specific mutations have become highly important as all cell types can be differentiated from iPSCs. We have generated and characterized two iPSC lines from patients-derived fibroblasts with defects in the PCCA and PCCB genes. These iPSC lines can be differentiated into cardiomyocytes that mimic the tissue-specific hallmarks of the disease. The presence of cardiomyocytes has been easily established by visual observation of spontaneously contracting regions, and the expression of several cardiac markers. PCCA iPSC-derived cardiomyocytes exhibited an alteration of autophagy process with an accumulation of residual bodies and mitochondrial dysfunction characterized by reduced oxygen consumption and alteration of mitochondrial biogenesis due to a deregulation of PPARGC1A. We also evaluated the expression of heart-enriched miRNAs previously associated with cardiac dysfunction and several miRNAs were found deregulated. Furthermore, we found increased protein levels of Herp, Grp78, Grp75, sigma-1R and Mfn2 suggesting ER stress and calcium perturbations in these cells.
We are planning to analyze PCCB cardiomyocytes to compare the results with PCCA and control data. We are working to obtain mature cardiomyocytes in order to perform electrophysiology studies (K+ currents) using a whole-cell patch clamp method. We are interested in the study of the tissue-specific bioenergetic signature comparing cardiomyocytes derived from control and PA patients´ iPSCs by reverse phase protein microarrays (RPPMA). Future work also includes testing the effect of the mitochondrial biogenesis activator, MIN-102 compound (PPAR agonist, derivative of pioglitazone) and of the mitochondrial targeting antioxidant MitoQ in PA cardiomyocytes.
We would like to sincerely thank the Propionic Acidemia Foundation for supporting our research.