Mindfulness Meditation
An Ancient Tradition with Practical Application
Vicki Ancell Sheahen, MBA, CPC, CCC
Pathway Coaching
“We cannot stop the waves, but we can learn to surf” Jon Kabat-Zin
Meditation has taken its rightful place in western society. The benefits of meditation, from reducing stress, modulating hormonal functioning, especially of oxytocin and cortisol,and reducing the intensity and frequency of negative and chronic stress reactions are now being documented and used by medical doctors, medical clinics, such as Mayo, and many other professionals today.
Meditation strengthens our ability to cope with difficult emotional experiences and increase emotional wellbeing by mitigating negative thinking, including rumination. Meditation as a way of being teaches us to manage the “narrative” in our head and helps us become emotionally proactive rather than reactive.
Mindfulness Meditation is a method that focuses on our breathing, noting when our mind wanders, and gently returning our attention back to our breath. This focus on our breath, noting our wandering mind and returning to our breath is training our brain to be focused and present in our daily lives, what Dan Harris calls the “off the bench benefits” of meditation. The goal of meditation is not to empty our mind, which is impossible, but to focus on the present in spite the narrative in our head.
Basic Breath Meditation
In my practice, when working with new student meditators, I recommend practicing 5 to 10 minutes a day. I also recommend finding a group or meditation coach in your area to help you grow and refine your practice. Set a timer so you are not worried about the time; 5 minutes is a very good and doable start.
Read the instructions below
When comfortable -set your timer for 5 minutes and begin
Find a comfortable position in which to sit for this period. As you allow your eyes to gently close, tune into your body and make any minor adjustments. It can be helpful to remember our intentions of both ease and awareness. Sit in a way that feels comfortable but alert.
We’ll start with a few minutes of concentration practice, just to help our minds settle and arrive in our present time meditation experience.Take a cleansing breath in and feel how the breath awakens your senses. As you breath out, imagine breathing out any tension, stress, or anxiety.
Now allow your body to resume its natural breathing and see where in the body you can feel the breath. It may be in the stomach or abdomen, where you can feel the rising and falling as your body breathes. It might be in the chest, where you may notice the expansion and contraction as your body inhales and exhales. Perhaps it’s at the nostrils, where you can feel a slight tickle as the air comes in, and the subtle warmth as your body exhales.
You can pick one spot to stay with for this meditation practice. As you become a witness to your breathing, we will use “labeling the breath” as a technique to help you stay focused. As you breathe in with awareness say silently to yourself “in” and on exhaling, say silently to yourself “out”.Remember that labeling the breath is a tool to help build concentration and focus and is not a measurement of how good a meditator you are.
You will notice your mind wandering. When your mind wanders, and it always will, we are being offered an opportunity to cultivate mindfulness and concentration. Each time we notice our mind wandering, we’re strengthening our ability to recognize our experience. Each time we bring the mind back to the breath, we’re strengthening our ability to focus on an object in the present moment. Treat this as an opportunity rather than a problem, and return to your “in” breath.
Resources
10% Happier: Dan Harris
Meditation for Fidgety Skeptics: Dan Harris, Jeff Warren, Carlyle Adler
Wherever You Go There You Are – Jon Kabat-Zin
Arriving at your own Door: 108 Lessons in Mindfulness – Jon Kabat-Zinn
No Time Like The Present- Jack Kornfield
Meditation Apps
10% Happier
Calm
Simply Being

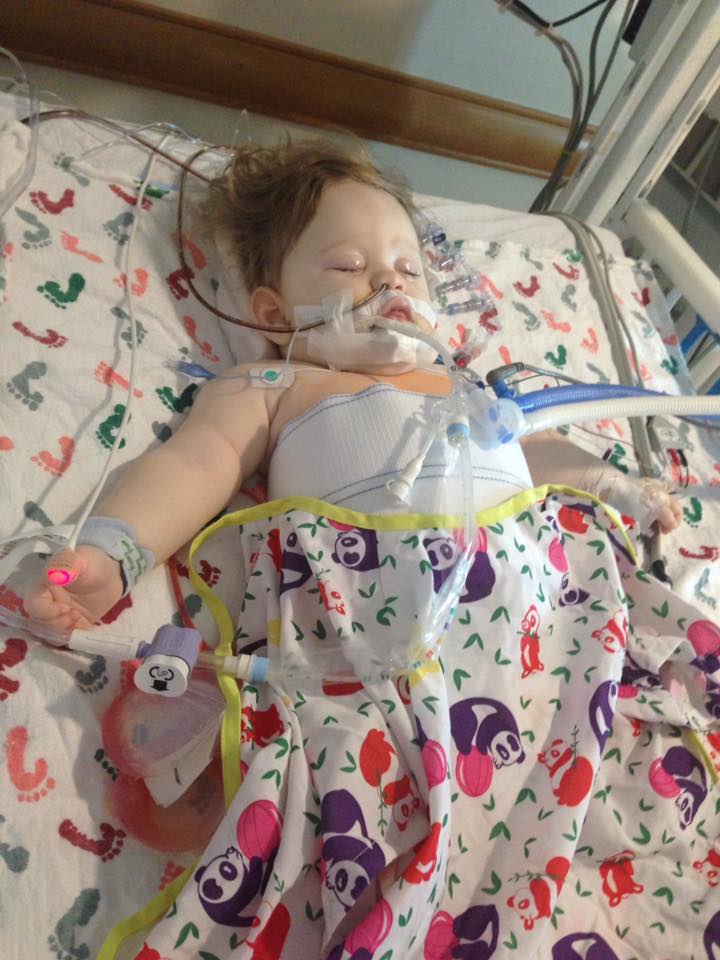
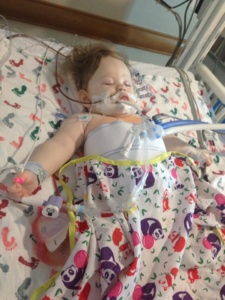 were very fortunate that our first call was “the call” that gave Annabelle her new liver. She went back for surgery around 10:30pm that night and they finished her surgery around 9am that next morning. After surgery Annabelle spent about one week in the PICU. After that week the transplant team moved herto the transplant recovery unit where she stayed until she was discharged. Around two weeks post-transplant Annabelle did encounter a small episode of rejection. Even though “rejection” sounds scary it is very common early on in transplant, and mild cases like Annabelle’s are generally treated with some high-powered IV steroids for a few days. Annabelle was discharged on August 30th and only spent a total of 21 days in the hospital. The transplant/genetics teams in Pittsburgh told us to prepare for complications (as is common with Organic Acidemia patients), but overall Annabelle had very few complications from her transplant surgery for which we are thankful.
were very fortunate that our first call was “the call” that gave Annabelle her new liver. She went back for surgery around 10:30pm that night and they finished her surgery around 9am that next morning. After surgery Annabelle spent about one week in the PICU. After that week the transplant team moved herto the transplant recovery unit where she stayed until she was discharged. Around two weeks post-transplant Annabelle did encounter a small episode of rejection. Even though “rejection” sounds scary it is very common early on in transplant, and mild cases like Annabelle’s are generally treated with some high-powered IV steroids for a few days. Annabelle was discharged on August 30th and only spent a total of 21 days in the hospital. The transplant/genetics teams in Pittsburgh told us to prepare for complications (as is common with Organic Acidemia patients), but overall Annabelle had very few complications from her transplant surgery for which we are thankful.














{kind=link}